



Коллектив авторов
Внутренние болезни. Том 2
5.4. АПЛАСТИЧЕСКИЕ АНЕМИИ
Под апластической анемией понимают такое состояние кроветворения, которое характеризуется наличием в крови панцитопении (анемии, лейкопении и тромбоцитопении) при разной степени выраженности гипоплазии или аплазии костного мозга. По природе своей она может быть врожденной или приобретенной, по генезу – миелотоксической или иммунной, а по течению – средней тяжести, тяжелой и сверхтяжелой. Апластические анемии встречаются с частотой 1 – 3,5 случая на 100 тыс. населения.
5.4.1. Врожденная апластическая анемия (Фанкони)
Примером врожденной апластической анемии может быть анемия Фанкони. Она наследуется по аутосомно-рецессивному типу и из-за нарушения синтеза ДНК сопровождается множественными случайными поломками хромосом, эндоредупликацией генома и хроматидными обменами.
Клиническая картина. Болезнь проявляет себя рано. В клинике, наряду с нарушением кроветворения и связанной с ним анемией, может быть разной степени выраженности геморрагический диатез, объяснимый тромбоцитопенией, а также повышенная чувствительность к инфекциям из-за гранулоцитопении. Это проявляется на фоне различных дефектов развития скелета (микроцефалия, отсутствие лучевой кости и больших пальцев кисти, маленький рост), врожденной гипоплазии почек, гипогонадизма, гиперпигментации кожи и т. д.
Наиболее серьезными осложнениями апластической анемии являются инфекционные осложнения и геморрагический диатез, которые сильнее выражены при тяжелой форме болезни, и в первую очередь требуется их коррекция.
Лабораторная диагностика. Анализ крови обнаруживает анемию, тромбоцитопению и гранулоцитопению разной степени выраженности с увеличением относительного содержания лимфоцитов и, возможно, плазматических клеток. Анемия носит явный гипорегенераторный характер с уменьшенным количеством ретикулоцитов.
Пунктат костного мозга скудный и малоклеточный. В трепанатах костномозговые фрагменты бедны кроветворными элементами, и более трех четвертей их замещены жировой тканью. В большинстве участков мало и гранулоцитарных, и эритроидных, и мегакариоцитарных элементов, а доминируют лимфоциты и плазматические клетки. Вместе с тем могут встречаться места нормальной клеточности с выраженными лимфоидными фолликулами.
Диагноз анемии Фанкони предполагается при наличии в клинике отмеченных выше соматических нарушений, которые сочетаются с развитием панцитопении крови. Он подтверждается данными трепанобиопсии костного мозга, а также результатами его цитогенетического исследования, которые выявляют аплазию кроветворения и характерные для анемии Фанкони множественные нарушения хромосом. Решающим для постановки диагноза является анализ хромосом после культивирования клеток с диэпоксибутаном или митомицином С, которые вызывают в этих клетках множественные индуцированные мутации.
Дифференциальный диагноз следует проводить с другими врожденными апластическими анемиями, в частности ассоциирующимися с врожденным дискератозом кожи, нарушением роста волос и ногтей. При этом могут быть еще телеангиэктазии на теле, наблюдаться резкое выпадение волос и нарушение потоотделения. У таких больных может быть и умственная отсталость, но хромосомные нарушения кроветворных элементов для них не характерны.
Лечение врожденной апластической анемии включает трансфузии эритроцитов и тромбоцитов и профилактическое введение десферала или эксиджада. В случае вторичной иммунизации больного на фоне гемотрансфузий могут быть использованы глюкокортикоиды и циклоспорин А. Некоторым больным может быть предложена спленэктомия. Однако радикальным способом лечения анемии Фанкони остается трансплантация костного мозга со всеми вытекающими из нее положительными и отрицательными последствиями.
Прогноз при этом заболевании тяжелый. Приблизительно у 20 % больных болезнь в течение 5 – 10 лет может трансформироваться сначала в МДС, а потом в острый нелимфобластный лейкоз.
5.4.2. Приобретенные апластические анемии
Выделяют две формы приобретенных апластических анемий: миелотоксическую и иммунную.
Этиология. Появлению миелотоксической формы приобретенной апластической анемии способствуют: радиация, цитостатики, инсектициды, мышьяк, органические растворители и ДДТ. В то же время иммунный механизм повреждения костномозгового кроветворения просматривается при действии на клетки таких лекарств, как левомицетин, сульфаниламиды, некоторые уросептики и соли золота. Кроме того, он явно присутствует при возникновении апластической анемии у больных гепатитами (А, В и С), перенесших вирусную инфекцию Эпштейна – Барр, у беременных и т. д.
Патогенез. В основе развития апластических анемий лежит резкое уменьшение в костном мозге количества полипотентных стволовых клеток и неспособность их или стромы обеспечить полноценный гемопоэз. В части случаев это происходит из-за прямого повреждающего действия на стволовые клетки неблагоприятных факторов внешней среды или цитостатиков, в части – через иммунные механизмы. В любом случае недостаточное количество в костном мозге полипотентных стволовых клеток приведет к недопродукции кроветворных элементов всех ростков и уровней созревания, к запустению костного мозга (см. цв. вкл., рис. 5.4) и к развитию панцитопении крови.
Клиническая картина. Клинические проявления апластической анемии могут быть различными. В некоторых случаях она возникает остро и быстро прогрессирует, в других – заболевание течет относительно спокойно. На первое место в клинической картине выступает анемический синдром. Больные жалуются на общую слабость, одышку, сердцебиение и на плохую переносимость физических нагрузок. Углубление тромбоцитопении может привести к подкожным кровоизлияниям, кровоточивости десен, меноррагиям и повторным кровотечениям со стороны слизистых оболочек. Проявлением глубокой нейтропении могут быть рецидивирующие инфекции, включая ангину, стоматит, пневмонию, парапроктит.
В анализе крови выявляется нормохромная анемия разной степени выраженности и сниженное количество ретикулоцитов. Количество лейкоцитов ниже нормы за счет уменьшения числа гранулоцитов. В тяжелых случаях может быть агранулоцитоз, абсолютная моноцитопения и лимфопения. Количество тромбоцитов снижено, в тяжелых случаях до 20 % 109 и ниже.
Диагноз. Мысль об апластической анемии должна возникать у врача при наличии у больного клинических проявлений недостаточности эритроидного (анемия), мегакариоцитарно-тромбоцитарного (геморрагический диатез) и гранулоцитарного (инфекции) ростков кроветворения, а также отмеченных выше изменений крови. Поскольку пунктат костного мозга у многих больных апластическими анемиями получить не удается, адекватным методом диагностики считается трепанобиопсия. Последняя выявляет выраженную аплазию кроветворения с замещением большей части костных фрагментов (> 75 %) жировой тканью. В большинстве анализируемых участков эритроидные, гранулоцитарные и мегакариоцитарные элементы отсутствуют, а встречаются в основном лимфоциты и плазматические клетки. В то же время отдельные фрагменты костного мозга больных апластическими анемиями могут быть достаточно клеточными и содержать лимфоидные фолликулы. При этом считается, что основными критериями для диагноза тяжелой формы апластической анемии являются: количество гранулоцитов менее 0,5 % 109/л; количество тромбоцитов менее 20 % 109/л; скорригированное на нормальные цифры эритроцитов количество ретикулоцитов менее 1 % при клеточности костного мозга менее 30 %.
Дифференциальный диагноз апластической анемии следует проводить с другими видами анемий, в первую очередь с В12– и фолиеводефицитными, миелодиспластическим синдромом, острым лейкозом и метастазами рака в костный мозг. В отличие от апластической анемии, при В12– и фолиеводефицитных анемиях в большинстве случаев миелодиспластического синдрома и острого лейкоза костный мозг богат клеточными элементами. Кроме того, в нем представлены в увеличенном количестве бластные элементы. В случае же аплазии костного мозга из-за метастазирования туда раковых клеток их пласты в трепанатах костного мозга обнаруживаются без большого труда.
Лечение приобретенной апластической анемии представляет собой сложную задачу. Оно начинается с изъятия у больного опасных в отношении аплазии костного мозга лекарств и защиты его от действия других потенциальных индукторов аплазии. По мере необходимости проводится заместительная терапия (трансфузии эритроцитарной массы и тромбоконцентрата). Осуществляется профилактика и лечение инфекционных и грибковых осложнений соответственно антибиотиками, в том числе неадсорбируемыми в кишечнике, и противогрибковыми препаратами (низорал, дифлюкан, нистатин, вифенд, каспофунгин).
Специальные методы лечения тяжелой идиопатической апластической анемии включают: а) антилимфоцитарный глобулин (АЛГ); б) большие дозы глюкокортикоидов; в) циклоспорин-А или сандимун; г) гемопоэтические факторы роста (эритропоэтин, гранулоцитарно-макрофагальный и др.); д) циклофосфан; е) трансплантацию костного мозга.
Назначение АЛГ позволяет получить эффект у 50 – 60 % больных. Чаще всего АЛГ применяют в сочетании с большими дозами метилпреднизолона, который, помимо усиления самой терапии, помогает ослабить такие побочные эффекты антилимфоцитарного глобулина, как лихорадка, гипотензия, сывороточная болезнь и некоторые другие. Возможно также комбинированное лечение тяжелой формы апластической анемии АЛГ, высокими дозами метилпреднизолона и сандимуна, которое, по некоторым статистикам, может быть эффективно у 70 – 80 % больных. Наконец, у некоторых больных с иммунными формами апластической анемии терапию АЛГ можно успешно комбинировать с назначением циклофосфана.
Альтернативным методом лечения апластической анемии может быть пульс-терапия метилпреднизолоном (1,0 г/сут) в течение 3 – 5 дней. Радикальным же методом лечения тяжелой формы апластической анемии являются миелотрансплантации. После трансплантации пятилетняя выживаемость наблюдается у 60 – 70 % больных.
Для лечения апластической анемии средней тяжести применяют глюкокортикоиды, например преднизолон в дозе 1 мг/кг/сут в течение 1 – 2 мес. с последующим постепенным снижением дозы, и андрогены (оксиметалон 2,5 мг/кг/сут). При отсутствии эффекта показана спленэктомия.
5.5. ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
Гемолитические анемии (ГА) представляют крайне разнородную группу заболеваний крови, при которых укорочена продолжительность жизни эритроцитов. По своей природе они могут быть врожденными и приобретенными.
Преждевременное разрушение эритроцитов может происходить двумя путями. Первый, более часто встречающийся, – внесосудистый или внутриклеточный гемолиз, при котором эритроциты разрушаются макрофагами ретикулоэндотелиальной системы. Как правило, он происходит при наличии на поверхности эритроцитов антител, к которым макрофаги проявляют особый интерес.
Другой причиной внутриклеточного гемолиза может быть повышенная врожденная или приобретенная деформабельность эритроцитов, затрудняющая их прохождение через синусоиды селезенки и, отсюда, ухудшающая их выживание. В случае внутрисосудистого гемолиза разрушение эритроцитов происходит непосредственно в сосудистом русле, а их содержимое поступает прямо в кровь. Как правило, повышенное разрушение эритроцитов сопровождается компенсаторным усилением их продукции, нередко в 6 – 8 раз.
5.5.1. Врожденные гемолитические анемии
В эту группу ГА входят анемии с нарушением мембраны эритроцитов (эритроцитопатии), дефектом ферментов (ферментопатии) и дефектом гемоглобина (гемоглобинопатии).
Наследственный микросфероцитоз. Врожденная микросфероцитарная гемолитическая анемия представляет собой группу семейных заболеваний, обусловленных патологией мембраны эритроцитов. У трех четвертей больных заболевание передается по аутосомно-доминантному типу, у четверти – по аутосомно-рецессивному.
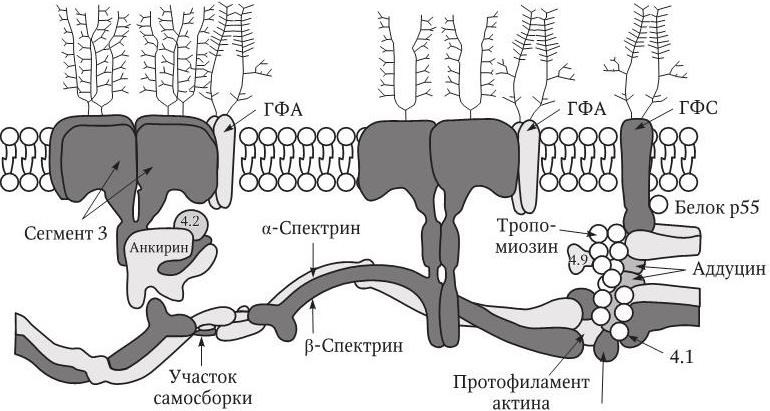
Рис. 5.5. Схематическое представление структуры цитоскелета эритроцита человека (Blood. Principles and Practice of Hematology. – Philadelphia: Lippincott, 1995. – Р. 1726): ГФА – гликофорин А; ГФС – гликофорин С
Патология эритроцитов при наследственном микросфероцитозе обусловлена дефектами генов основных мембранных белков цитоскелета эритроцитов (рис. 5.5), а именно спектрина, гены á и â которого расположены в локусах хромосом 1q22-q23 и 14q23-q24.2 соответственно; анкирина (8р11.2 – 70 %), АЕ1 – обменника анионов (17q21-qter – 25 %) или белка 4.2 (15q15). Необходимо отметить, что, несмотря на отмеченные генетические дефекты и аномальные цитоскелетные белки, костный мозг этих больных продуцирует эритроциты нормальной формы. Однако ввиду того что часть мембраны не поддерживается белками цитоскелета, при прохождении синусов селезенки она утрачивается. В результате отношение площади эритроцита к его объему постепенно уменьшается. Клетки становятся сферическими (см. цв. вкл., рис. 5.6), а размеры их отчетливо уменьшаются. Общая деформабельность таких эритроцитов еще больше снижается. В итоге они не могут пройти в отверстия селезеночных синусов, захватываются макрофагами и разрушаются.
Клиническая картина. Клиническая симптоматика наследственного микросфероцитоза включает анемический синдром, желтуху, изменение костей скелета и спленомегалию. Анемический синдром, как правило, выражен умеренно. Проявление желтухи варьирует и во многом зависит от функционального состояния печени. Если она работает хорошо, желтухи может не быть даже при выраженном гемолизе. В случае же функциональной недостаточности печени даже умеренный гемолиз может сопровождаться выраженной желтухой. Почти у всех больных имеет место спленомегалия разной степени выраженности. Чрезвычайно характерны также такие изменения костей, как «башенный череп» и «высокое нёбо», которые являются прямым отражением ненормального развития плоских костей новорожденных в условиях резко активированного гемолизом эритропоэза. Основными осложнениями заболевания могут быть гемолитические и апластические кризы, а также желчнокаменная болезнь.
Диагноз заболевания ставится при наличии у больного повторяющейся в течение жизни желтухи, отмеченных выше изменений со стороны костной системы и спленомегалии при выявлении подобных симптомов у нескольких членов семьи. Подтверждается диагноз обнаружением в крови микросфероцитов (менее 6 μ в диаметре), увеличенным содержанием ретикулоцитов, активацией эритроидного ростка в костном мозге, выявлением укороченной длительности жизни эритроцитов после мечения их Сr51. Осмотическая стойкость эритроцитов при наследственном микросфероцитозе может быть снижена, хотя у некоторых больных это становится очевидным только после суточной инкубации эритроцитов при температуре 37 °C. С другой стороны, спонтанный лизис эритроцитов после 48-часовой их инкубации по сравнению с контролем может быть увеличен во много раз. Содержание гемоглобина крови обычно превышает 90 г/л, но в период кризов может резко снижаться. При обострении может быть отмечено повышение в крови непрямого билирубина, увеличенное содержание в кале стеркобилиногена и стеркобилина, а в моче – уробилиногена и уробилина. При ультразвуковом исследовании печени может быть выявлен холелитиаз.
Дифференциальный диагноз микросфероцитарной анемии следует проводить с другими гемолитическими и негемолитическими анемиями и различными видами желтух, в том числе доброкачественных. В отличие от других гемолитических анемий, диагноз микросфероцитарной анемии ставится с учетом наследственной природы заболевания, возможности характерных изменений плоских костей и выраженности феномена микросфероцитоза.
Отличить микросфероцитарную анемию от негемолитических желтух помогает выраженный микросфероцитоз и ретикулоцитоз в крови, снижение осмотической стойкости эритроцитов, наличие в костном мозге компенсаторной реакции эритроидного ростка на гемолиз, а также обнаружение спленомегалии без признаков портальной гипертензии. В трудных случаях могут помочь молекулярно-биологические исследования генов.
Лечение. Единственным способом лечения микросфероцитарной анемии является спленэктомия.
Наследственный эллиптоцитоз. Другим видом врожденной эритроцитопатии является эллиптоцитоз (см. цв. вкл., рис. 5.7).
В основе заболевания лежат мутации тех же генов á и â цепей спектрина, расположенных соответственно в локусах хромосом 1 (1q22-q23) и 14 (14q23-q24.2), а также белка 4.2 и гликофорина С, гены которых расположены в локусах хромосом 15 (15q15) и 2 (2q14-q21) соответственно. Что касается окончательных причин различия в форме эритроцитов при микросфероцитозе и эллиптоцитозе, они до конца не ясны.
Клиническая картина, принципы диагностики и терапии напоминают таковые при наследственном микросфероцитозе.
Гемолитическая анемия, связанная с дефицитом в эритроцитах глюкозо-6-фосфат-дегидрогеназы. В силу особенностей структуры эритроцита человека, не содержащего ни ядра, ни митохондрий, ни рибосом, единственным источником его энергии является глюкоза. Она диффундирует в эритроцит из плазмы крови и превращается в глюкозо-6-фосфат. Подавляющая часть глюкозо-6-фосфата (до 90 %) используется в ходе анаэробного гликолиза, остальной – с помощью пентозофосфатного шунта. Основным ферментом, участвующим в превращении глюкозы и энергоснабжении эритроцита, является глюкозо-6-фосфат-дегидрогеназа (Г6ФД), которая необходима для синтеза никотинамиддинуклеотидфосфата и глютатиона. В итоге при недостатке данного фермента в эритроците он недополучает и никотинамиддинуклеотидфосфат, и глютатион. В то же время известно, что восстановление глютатиона является основным механизмом защиты гемоглобина и мембран эритроцитов от окисления и, следовательно, от разрушения под действием многих окисляющих веществ, в том числе лекарств.
Дефицит Г6ФД связан с Х-хромосомой и поэтому поражает в основном мужчин. Он зарегистрирован приблизительно у 200 млн человек, живущих в основном в Средиземноморье, на Ближнем Востоке, в Западной Африке и Юго-Западной Азии.
Клиническая картина. Дефицит Г6ФД обнаруживает себя в клинике острым внутрисосудистым гемолизом, который часто возникает на фоне острых инфекционных или каких-то других заболеваний, а также после приема различных медикаментов или еды конских бобов. Клинические проявления гемолиза обычно обнаруживают себя на 2 – 5-й день после воздействия провоцирующего агента. Выраженность симптомов может быть различной – от легкой желтушности до острой почечной недостаточности. Главными же признаками внутрисосудистого гемолиза будут лихорадка (как при малярии), боли в пояснице, появление темной или черной мочи из-за присутствия в ней гемоглобина.
Лабораторная диагностика. В межкризовый период картина крови не изменена. В период криза выявляется анемия, иногда очень тяжелая, с фрагментированными эритроцитами, тельцами Гейнца (окисленный денатурированный гемоглобин) (см. цв. вкл., рис. 5.8) в ретикулоцитах, повышенным ретикулоцитозом. При биохимическом исследовании в крови находят свободный гемоглобин, уменьшение содержания связывающего свободный гемоглобин гаптоглобина и умеренное повышение непрямого билирубина. В моче также обнаруживают свободный гемоглобин, а при умеренном гемолизе и наличии клеточного осадка – гемосидерин. В связи с тем, что количество Г6ФД в ретикулоцитах и молодых эритроцитах выше, чем в зрелых, дефицит этого фермента у больного может быть временно замаскирован. Однако повторное определение Г6ФД после купирования криза выявляет его дефицит.
Диагноз Г6ФД-дефицитной гемолитической анемии ставится на основе характерных особенностей гемолиза. В частности, появления на фоне инфекции или приема лекарств немотивированно высокой температуры тела, болей в пояснице, гемоглобинемии, гемоглобинурии и характерной для нее темной мочи, а также снижения гаптоглобина крови. Окончательный диагноз ставится после выявления у больного дефицита Г6ФД.
Дифференциальный диагноз этой формы гемолитической анемии проводится с пароксизмальной ночной гемоглобинурией (болезнь Маркиафава – Микели) и гемолизиновой формой приобретенной гемолитической анемии (см. ниже).
Лечение показано только при гемолитическом кризе. Оно начинается с немедленной отмены вызвавшего гемолиз препарата (делагил, фенацетин, аспирин в больших дозах, сульфаниламиды, нитрофураны, ПАСК и т. д.). Проводится инфузионное введение физиологического раствора, направленное на профилактику острой почечной недостаточности. В тяжелых случаях проводят обменные переливания крови и методы экстракорпоральной детоксикации. Трансфузия эритроцитарной массы показана только в случае тяжелой анемии. При этом в качестве доноров не должны использоваться родственники больного, у которых также может иметь место дефицит Г6ФД.
Профилактика гемолитических кризов заключается в своевременной диагностике заболевания, в том числе у родственников больного, тщательном опросе больных перед дачей лекарств о наличии гемолитических кризов в анамнезе, а также в объяснении больным необходимости осторожного обращения с лекарствами в будущем.
Гемоглобинопатии – это группа гемолитических анемий, обусловленных врожденным дефектом структуры глобина. Описано около 200 клинически значимых гемоглобинопатий, в основе которых лежат мутации глобина. Однако только некоторые из них сопровождаются развитием анемического синдрома.
Этиопатогенез. В норме кровь взрослых людей содержит три типа гемоглобина. Основным является гемоглобин А (Hb A), состоящий из двух á-цепей и двух â-цепей глобина (α2β2). На него приходится 96 – 98 % всего гемоглобина (см. цв. вкл., рис. 5.9).
В гемоглобинах F и А2, которые составляют соответственно 1 – 2 % или 2 – 3 % от общего содержания гемоглобина, β-цепь глобина заменяет γ-цепь (Hb F – α2γ2) или δ-цепь (Hb A2 – α2δ2). Синтез каждой цепи глобина кодируется отдельными генами, которые находятся на хромосомах 11 и 16. Наиболее часто встречающиеся гемоглобинопатии (S,CиЕ)обусловлены аномалией β-цепей глобина, а α-цепь при этом нормальная. Значительно реже встречается наследование двух различных патологических вариантов β-цепи глобина, по одному от каждого из родителей – двойная гетерозиготная гемоглобинопатия, примером которой является гемоглобинопатия SC. Среди гемоглобинопатий, связанных с Hb S, только гомозиготные или двойные гетерозиготные варианты обнаруживают выраженные клинические проявления. Напротив, нестабильные подварианты гемоглобина встречаются только при гетерозиготных вариантах, поскольку гомозиготные обычно несовместимы с жизнью.
Структурные особенности примерно 90 % аномальных гемоглобинов заключаются в замещении одной аминокислоты. Хорошей иллюстрацией гемоглобинопатии может быть серповидно-клеточная анемия.
Серповидно-клеточная анемия (СКА) – это врожденная гемолитическая анемия, обусловленная носительством гемоглобина с измененной структурой, получившего название Hb S.
Эпидемиология. СКА была описана Herrick в 1910 г. Она встречается во всех регионах мира, хотя чаще в неблагополучной по малярии Африке. В Европе больше всего носителей Hb S приходится на жителей Средиземноморья и Кавказа. С другой стороны, в силу сложившихся исторических особенностей формирования населения в США СКА с 72 000 зарегистрированных больных среди наследственных патологий стоит на первом месте.
Патогенез. Суть заболевания состоит в точечной мутации в 6-м кодоне гена ââ-цепи глобина, следствием чего является замещение глютаминовой кислоты на валин. Включение âs-цепи в тетрамер приводит к образованию Hb S. Нерастворимость деоксигенированного Hb S приводит к его полимеризации и, как следствие, к снижению деформабельности и образованию серповидных эритроцитов (см. цв. вкл., рис. 5.10). В основе заболевания лежит точечная мутация в гене ââ-цепи глобина, расположенного на хромосоме 11 в локусе 11р15.4. В результате точечной GAG D GTG мутации в 6-м кодоне â-цепи глобина формирующаяся в результате трансляции â-цепь глобина получает вместо глутаминовой кислоты валин. В свою очередь, включение таких âs-цепей в состав тетрамера глобина приводит к тому, что из-за плохой растворимости Hb S в условиях деоксигенации или высокой концентрации гемоглобина он полимеризуется. Это приводит к нарушению естественной деформабельности эритроцитов и к их серповидноклеточности (см. цв. вкл., рис. 5.11). Естественно, что, проходя по сосудистому руслу и селезенке, такие эритроциты повреждаются, разрушаются и, более того, ответственны за закупорку части мелких сосудов и ишемию снабжаемых ими органов и тканей.
Клиническая картина. Клинические проявления СКА вариабельны и более выражены при гомозиготном (Hb SS), чем при гетерозиготном (Hb SC) варианте. Обострение заболевания носит кризовый характер. При этом часть больных в межкризовый период чувствуют себя хорошо, в то время как крайне тяжелые варианты могут закончиться летальным исходом еще в раннем детском возрасте. В качестве провоцирующих кризы факторов выступают: 1) инфекции; 2) стресс; 3) обезвоживание и увеличение осмолярности плазмы; 4) снижение парциального давления О2 в крови; 5) холод; 6) применение сосудосуживающих препаратов; 7) некоторые другие реже встречающиеся причины. Как правило, заболевание себя не проявляет до тех пор, пока не произойдет замена Hb F на Hb S, что обычно имеет место через полгода. Ведущее место в клинике занимают периодически возникающие боли в различных участках тела и хроническая гемолитическая анемия, протекающая по типу кризов с внесосудистым компонентом гемолиза. Из других проявлений характерны тяжелые инфекции, как правило, начинающиеся уже в раннем детстве. Кроме гемолитических кризов возможно развитие апластических кризов. Серповидноклеточный криз характеризуется появлением острых болей в костях, спине, животе, конечностях, в области печени и/или селезенки. Как уже говорилось, болевой синдром связан с окклюзией сосудов и развитием инфарктов в тканях, на фоне которых нередко имеет место присоединение органной и полиорганной недостаточности. В частности, та же причина лежит в основе повышенной склонности к инфекциям, когда из-за множественных тромбозов сосудов и синусов селезенки развивается функциональная аспления. Наконец, такой же генез свойственен и неврологическим осложнениям (ишемические и геморрагические инсульты с развитием парезов, параличей, судорог, головных болей, рвоты, фотофобии). Здесь необходимо подчеркнуть, что у больных СКА могут быть немые крупные множественные инфаркты мозга, которые могут приводить к необъяснимому другими причинами снижению интеллекта и памяти.
Крайне характерно, хоть и вариабельно, при СКА поражение паренхимы печени, желчевыводящей системы и почек. Так, желчнокаменная болезнь проявляется уже в детском возрасте. Как вариант серповидно-клеточного криза часто развивается острый печеночный криз с некрозом гепатоцитов, который проявляет себя гепатомегалией, нарастанием желтухи, лихорадкой, увеличением АСТ и билирубина. Как правило, длительность печеночного криза не превышает двух недель, но может индуцировать печеночную недостаточность. Другим вариантом печеночного криза у больных СКА является острый печеночный криз с секвестрацией, который проявляется быстрым увеличением размеров печени и падением уровня гемоглобина в крови. Следующим вариантом печеночного криза может быть острый печеночный криз с внутрипеченочным холестазом, который проявляется выраженной гипербилирубинемией (без увеличения температуры), болями в печени, лейкоцитозом, печеночной недостаточностью и в конечном итоге приводит к смерти. Кроме того, в генезе поражения печени у больных СКА могут участвовать также вирусный гепатит или аутоиммунный гепатит и гемосидероз.
Поражение почечных сосудов и клубочков (см. цв. вкл., рис. 5.10) сопровождается развитием острой или хронической почечной недостаточности. При этом как во время гемолитического криза, так и длительное время после него могут иметь место: гипостенурия, протеинурия, гематурия и снижение клубочковой фильтрации. Неудивительно, что в этих условиях довольно часто развивается вторичная артериальная гипертензия.
Далее больным СКА свойственно поражение сердца и легких, в частности развитие острого инфаркта миокарда или ишемической кардиомиопатии с присоединением тяжелой сердечной недостаточности. В свою очередь, нарушение микроциркуляции в легких и тромбозы легочных сосудов предрасполагают к развитию отека легких и тяжелой легочной гипертензии. Из других проявлений заболевания следует упомянуть о: а) частом снижении зрения, связанном с развитием ретинопатии; б) некрозах и язвенных поражениях кожи; в) возможности изменения конфигурации костей черепа и замедления физического и психического развития пациентов.
Для женщин с СКА характерны дисменорея, поликистоз яичников, фиброматоз молочных желез, а в случае беременности – высокий риск летальных осложнений как для матери, так и для плода, а для мужчин – приапизм, импотенция и, реже, гипогонадизм.
Лабораторные данные. Гипохромные микроцитарные дискоциты в мазках крови свойственны Hb SC, нормохромные эритроциты – Hb SS- и Hb S+ â-талассемиям. При всех вариантах СКА встречается пойкилоцитоз. Характерно также снижение гематокрита, числа эритроцитов и показателей гемоглобина крови как в межкризовый период, так и в период криза. Практически у всех больных имеет место ретикулоцитоз. Он меньше выражен при Hb S+ â-талассемия подварианте, а больше – в период гемолитического криза. Характерно появление Howell-Jolly телец в эритроцитах. В то же время только Hb SS-подварианту заболевания свойственны повышенный лейкоцитоз и тромбоцитоз. Наконец, характерные для СКА увеличение неконъюгированного билирубина, лактатдегидрогеназы, снижение гаптоглобина отражают степень гемолиза.
Диагноз. Мысль о СКА возникает у врача при наличии у больных признаков хронической гемолитической анемии врожденного характера, протекающей на фоне тяжелых инфекций и сопровождающейся ишемическими болями различной локализации. Подтверждает диагноз наличие в мазках крови серповидно-клеточных эритроцитов, а также электрофоретические и молекулярно-биологические исследования, направленные на предмет выявления Hb S. Что касается разграничения различных подвариантов заболевания, в частности Hb SS, Hb SC или Hb S+ â-талассемия, для достижения этой цели необходимо проведение специальных исследований. Среди них: а) электрофорез гемоглобина на целлюлозоацетатной пленке при рН 8,4; б) электрофорез гемоглобина на цитратном агаре при рН 6,2; в) тест на растворимость гемоглобина; г) ДНК-типирование (ПЦР, рестрикционный анализ, аллель-специфическая гибридизация и др.). В меньшей мере разграничению подвариантов СКА помогает анализ морфологии эритроцитов. В частности, серповидные эритроциты имеют место при гомозиготной (Hb SS) СКА, а мишеневидные эритроциты – при сочетании СКА с â-талассемией.