



А. С. Брюховецкий
Персонализированная медицина нового поколения: клональная реконституция гемопоэза и CHIP-основанная
Глава 2. Клональный гемопоэз – фундаментальная причина возникновения и прогрессирования большинства болезней цивилизации и старения
Что такое термин «гемопоэз», хорошо известно врачам и биологам. Гемопоэз (от др.-греч. αἷμα – кровь и ποίησις – выработка, образование, от ποιεῖν – творить), кроветворение – это процесс образования, развития и созревания клеток крови: лейкоцитов, эритроцитов, тромбоцитов у позвоночных («Википедия», 2024). Под этим термином также понимается весь комплекс органов и тканей системы кроветворения человека и млекопитающих. Классифицируют эмбриональный (внутриутробный) гемопоэз и постэмбриональный гемопоэз. Но в этой главе мы хотели бы обсудить проблему клонального гемопоэза, так как считаем его фундаментальной причиной возникновения и прогрессирования основных болезней цивилизации (БЦ) и главным элементом патогенеза этих болезней (Брюховецкий А. С., Шурдов М. А., 2023). Клональный гемопоэз (КГ) характеризуется наличием популяции клеток крови, происходящих из мутировавших мультипотентных стволовых клеток/клеток-предшественников, которые получили преимущество в аномальном росте (Cacic A. M., Schulz F. I., Germing U. et al., 2023). Это определение КГ, данное исследователями из Дрезденского университета, очень емкое и системное и отражает суть процесса. Как утверждают в журнале Blood в своем научном обзоре по клональному гемопоэзу G. A. Challen and М. A. Goodell (2022), недавнее открытие распространенности клонального кроветворения (КГ) изменило представление врачей-гематологов и онкологов о гемопоэтических стволовых клетках (ГСК). На протяжении десятилетий было признано, что у нас есть множество ГСК, большинство из которых являются излишними и даны нам про запас, на всякий случай. Как правило, ГСК находятся в состоянии покоя, редко делятся, но иногда активируются для дифференцировки. Оценки общего количества ГСК в костном мозге человека варьируются от ∼10 000 (Catlin S. N., Guttorp P., Abkowitz J. L., 2005; Xu J., Wang Y., Guttorp P., Abkowitz J. L., 2018) до ∼200 000 (Lee-Six H., Øbro N. F., Shepherd M. S. et al., 2018; Watson C. J., Papula A. L., Poon GYP et al., 2020). На протяжении всей жизни все соматические клетки непрерывно накапливают мутации, и ГСК не являются исключением, приобретая около 10 мутаций в год (1—2 на клеточное деление; рисунок 1) (Welch J. S., Ley T. J., Link D. C. et al., 2012). Таким образом, к зрелому возрасту (приблизительно 20 лет) большинство наших ГСК будут содержать две кодирующие мутации и 200 некодирующих мутаций (которые также могут влиять на функцию). Если у нас есть 100 000 ГСК, этот 20-летний ребенок накопит порядка 200 000 кодирующих мутаций в примерно 20 000 генов, разбросанных по всему пулу стволовых клеток, что делает все ГСК немного разными. Количество поражений на ген продолжает линейно увеличиваться с возрастом (Welch J. S., Ley T. J., Link D. C. et al., 2012).

Рис. 1. Схема развития клонального гемопоэза (КГ). При рождении пул ГСК относительно однороден. Со временем соматические мутации
накапливаются со скоростью примерно 10 в год, так что все ГСК немного отличаются у юношества (ранних взрослых). Эти различия проявляются в недостатках и преимуществах для выживания и участия в производстве периферической крови, в результате чего некоторые ГСК «побеждают» с возрастом (эритроциты справа). В то время как расширенные клоны
могут быть обнаружены в среднем возрасте с помощью чувствительных методов секвенирования. В настоящее время принято определение, когда клон достигает доли 4% клеток, измеренных в периферической крови.
Это соответствует частоте вариантного аллеля (VAF) 2%, когда варианты
(мутации) являются гетерозиготными
(Цит. по: Challen G. A. and Goodell M. A., 2020)
Хотя точное количество ГСК у взрослых до сих пор неясно (Xu J., Wang Y., Guttorp P., Abkowitz J. L., 2018), каждый ген получит несколько «попаданий» в течение жизни (в зависимости от размера гена и состава кодонов) (Watson C. J., Papula A. L., Poon GYP et al., 2020), а общее количество кодирующих мутаций распространяется по всей стволовой клетке. Количество мутаций поразительно (потенциально порядка 1 млн к 70 годам). В обзоре S. Jaiswal (2020) показано, что количество мутаций к 70 годам составляет от 350 000 до 1,4 млн нуклеотидных замен.
Эти мутации делают каждую ГСК уникальным «бегуном» с немного отличающимися характеристиками, которые могут помочь или снизить его шансы на победу с течением времени. Считается, что КГ развивается с возрастом в течение очень длительного периода времени, аналогично марафонскому забегу. Подобные ассоциации пришли по поводу КГ к G. A. Challen and M. A. Goodell (2020), которые для иллюстрации и лучшего понимания проводят аналогию КГ на примере марафона. «В марафоне, который начинается с изначально хорошо подобранных бегунов, небольшое преимущество в выносливости может окупиться, в то время как бегун, который тратит большое количество энергии на ранней стадии, может не продержаться долго. Точно так же бегуны, получившие травмы, выбывают из игры. В долгой гонке в игру могут вступить многие второстепенные факторы, включая психологию, погоду и местность. Шанс всегда играет определенную роль, и, наконец, вероятность победы также зависит от количества соревнующихся бегунов». Сочетание общего количества накопленных мутаций в нашей большой продолжительности жизни и исходного размера пула ГСК объясняет неизбежность КГ, которую можно наблюдать с помощью глубокого секвенирования даже в относительно молодом возрасте (Young A. L., Challen G. A., Birmann B. M., Druley T. E., 2012). Хотя флуктуация активности клонов стволовых клеток была оценена давно, в целом вклад стволовых клеток в выработку крови считался довольно стабильным при отсутствии явных заболеваний, таких как лейкемия или недостаточность костного мозга. По сути, КГ является результатом конкуренции долгоживущих стволовых клеток (СК) в костном мозге (КМ). Когда мы экстраполируем эти концепции на КГ, мы задаемся вопросом: как стволовая клетка может «победить»?
G. A. Challen and М. A. Goodell (2022) основное внимание уделили предполагаемым механизмам, которые приводят к КГ, особенно в контексте биологии стволовых клеток, на основе нашего текущего понимания функции некоторых генов, связанных с КГ. Они убеждены, что главная роль в механизмах формирования КГ принадлежит приобретенным дополнительным соматическим мутациям генов. Большинство приобретенных мутаций не имеют значения, а многие даже вредны для функции ГСК, что приводит к их гибели. Редкие мутации, дающие какие-либо преимущества ГСК, со временем увеличат его вероятность доминирования над конкурентами; однако только определенные виды преимуществ со временем проявятся в виде более крупного «клона» ГСК (рисунок 2). Наиболее мощными мутациями будут те, которые приводят к повышенному самообновлению ГСК, создавая больше ГСК, а не сбалансированную дифференцировку. Действительно, в обзоре 150 генов, в котором изучалось влияние нокаута гена (KГ) у мышей, большинство оказывало вредное влияние на функцию ГСК, в то время как некоторые приводили к экспансии ГСК (Rossi L., Lin K. K., Boles N. C. et al., 2012). Интересно, что три из этих генов (DNMT3A, TET2 и Cbl), как позже было показано, периодически мутируют и формируют КГ. Поскольку требуется время для накопления достаточного количества мутаций, а затем для того, чтобы эти мутации показали биологическую экспансию у людей, КГ редко встречается в возрасте до 70 лет, если использовать классическое определение, когда пороговым значением является больше 2% клона VAF в крови. Эти пороги и их клиническая значимость обсуждаются далее в других главах монографии.

Рис. 2. Модель делений ГСК, которые могут привести к клональному
гемопоэзу. (А) Существует баланс между регенерацией стволовых клеток (синий) и дифференцированных клеток (оранжевый). Эта схема не обязательно подразумевает асимметричное деление, но изображает чистый
результат (пунктирная рамка). (В) Стволовая клетка делится быстрее, но каждое решение имеет тот же чистый результат, что и на панели А.
Следовательно, сеть после большого количества клеточных делений
не увеличивает пул стволовых клеток и не превосходит нормальные ГСК. (С) ГСК имеет небольшой уклон в сторону самообновления. Каждые несколько делений (красные стрелки) он создает дисбаланс, так что
со временем в результате образуется больше стволовых клеток.
Скорость, с которой это приводит к действительно предвзятым выводам, будет зависеть от частоты несбалансированных решений. Для большинства генов КГ это, вероятно, изначально очень тонкая предвзятость,
объясняющая очень долгий период времени, в течение которого КГ
становится очевидным (Цит. по: Challen G. A.and Goodell M. A., 2020)
Существует ограниченное количество генов, мутация которых увеличивает самообновление ГСК и проявляется в виде КГ (Рисунок 3). Необходимо было секвенировать достаточное количество людей преклонного возраста, чтобы исчерпывающе идентифицировать все потенциальные гены (Watson C. J., Papula A. L., Poon GYPet al., 2020), но основные игроки, безусловно, были идентифицированы.

Рис. 3. Наиболее часто мутирующие гены клональности и доля
распространенных мутаций клонального гемопоэза (КГ) у людей, которые можно идентифицировать как драйвер. Они варьируются от исследования к исследованию, но указаны общие пропорции (Цит. по: Challen G. A. and Goodell M. A., 2020)
Типы генов, которые рекуррентно мутируют при КГ, в целом делятся на три основных класса: эпигенетические регуляторы, факторы сплайсинга и гены ответа на повреждение ДНК (DDR), с более редким вкладом членов cohesin, сигнальных молекул и гемопоэтических факторов транскрипции.
Мышиные модели оказались полезными, чтобы начать анализ роли, которую играют мутации в этих генах. Генетические манипуляции со многими из этих генов приводят к повышению приспособленности и самообновлению ГСК. В соответствии с концепцией, что небольшие преимущества важны в феномене, который развивается в течение десятилетий, преимущество, обеспечиваемое этими мутациями, часто неуловимо, что приводит к незначительным изменениям в молекулярной и клеточной функции, что затрудняет механистическое объяснение того, как эти мутации работают.
Обобщая вышеизложенные соображения, американские исследователи Grant A. Challen and Margaret A. Goodell (2020) пришли к выводу, что в контексте биологии стволовых клеток и на основе нашего текущего понимания функции некоторых генов, связанных с КГ, нужно понимать то, что, хотя большинство мутаций не оказывают никакого влияния, некоторые из них приводят к тонким функциональным различиям, которые в конечном итоге проявляются в различном поведении стволовых клеток. При большом пуле стволовых клеток и многих десятилетиях конкуренции некоторые из этих различий дают преимущества в определенных условиях. Примерно 20 генов периодически обнаруживаются как мутированные при КГ, что указывает на то, что они дают некоторое преимущество. Влияние этих мутаций начали анализировать на молекулярном уровне путем моделирования на клеточных линиях и на мышах. Мутации в эпигенетических регуляторах, таких как DNMT3A и TET2, дают преимущество, усиливая самообновление стволовых клеток и клеток-предшественников и ингибируя их дифференцировку. Мутации в других генах, участвующих в реакции на повреждение ДНК, могут просто повысить выживаемость клеток.
На рисунке 4 представлены основные стратегии победы, которые используют ГСК для доминирования над другими ГСК костного мозга в организме. Один из путей доминирования ГСК – это стратегия их самообновления, молекулярно-биологической основой которой являются мутации в гене DNMT3A. Другой подход к доминированию среди других клонов кроветворения носит название «стратегии выживания» и обусловлен мутациями в гене ТЕТ2.
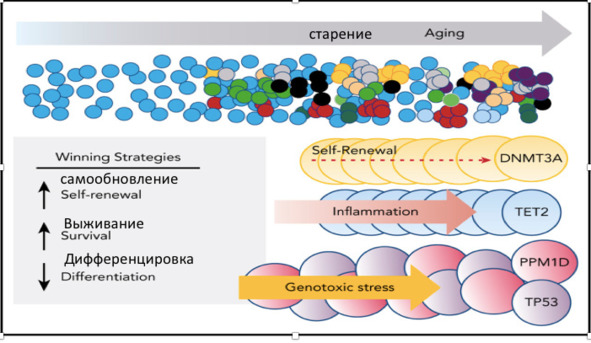
Рис. 4. Стратегии победы ГСК для формирования доминирования
определенных клонов кроветворных стволовых клеток
(Цит. по: Challen G. A. and M.A. Goodell М. А., 2020)
Наличие мутаций гена DNMT3A, кодирующего фермент ДНК-метилтрансферазу de novo, определяет ответственность за установление новых паттернов метилирования ДНК во время развития. Эти мутации свидетельствуют о нарушении принятия решений о судьбе стволовых клеток. Клетки крови, несущие мутации в гене DNMT3А, способны вызывать более сильный иммунный ответ как против нормальных тканей хозяина, так и против опухоли. Мутации в эпигенетических регуляторах, таких как DNMT3A и TET2, дают преимущество, усиливая самообновление стволовых клеток и клеток-предшественников и ингибируя их дифференцировку. Мутации в эпигенетических регуляторах доминируют в ландшафте КГ, при этом около 50% всех мутаций КГ являются вариантами DNMT3A. Большинство мутаций DNMT3A при КГ являются гетерозиготными и, вероятно, приводят к потере функции белка по разным механизмам. Мутации DNMT3A при КГ распространяются по всей длине гена, при этом миссенс-мутации группируются в известных структурных и функциональных доменах. Важно отметить, что спектр вариантов DNMT3A при КГ отличается от наблюдаемого в миелоидных новообразованиях, поскольку он заметно менее обогащен остатком горячей точки R882. Уровень активности DNMT3A, остающийся после мутации, фактически «дозировка» DNMT3A, будет коррелировать со скоростью размножения мутантных клонов. Клональный гемопоэз, ассоциированный с мутациями в этом гене, резко увеличивает риск сердечно-сосудистых заболеваний. Мутации TET2 в гемопоэтических стволовых клетках способствуют самообновлению ГСК, давая им конкурентное преимущество в плане экспансии по сравнению с другими клонами.
Другой подход к доминированию в кроветворении носит название «Стратегии выживания». Эта стратегия направлена на дифференцировку ГСК в результате генотоксического стресса. Эта стратегия обусловлена мутациями в гене ТР53. Соматические мутации в TP53 являются центральными эффекторами реакции на повреждение ДНК, вызванной незащищенными концами теломер. Они повышают вероятность того, что истощение теломер в популяции ГСК отбирает клетки с генетической способностью избегать путей старения без необходимости репарации лежащих в основе дефектов удлинения теломер. Соматические мутации ТР53 опосредуют миелоидную трансформацию при СШД. В исследовании SDS Registry наличие, количество и распространенность аллелей соматического гена TP53 и мутации в нем не были предикторами неминуемого риска лейкемии у пациентов с СШД. Вместо этого прогрессирование мутантных по TP53 клонов было опосредовано развитием биаллельных изменений в локусе TP53, делецией, потерей гетерозиготности с нейтральной копией или второй точечной мутацией. В функциональных экспериментах было показано, что инактивация TP53 повышает конкурентоспособность SBDS-дефицитных клеток за счет инактивации путей старения без коррекции лежащих в основе SDS рибосом и дефектов трансляции. В соответствии с этими генетическими наблюдениями человека потеря TP53 может разъединить рибосомный стресс, вызванный дефицитом SBDS, от активации путей клеточного старения, что приводит к частичному восстановлению фенотипа СШД. Инактивация TP53 ограничивает клеточные последствия неэффективности трансляции и дефектной длины теломер, соответственно указывая на то, что активация пути p53 ограничивает относительную приспособленность и опосредует устранение «неудачников». Таким образом, инактивация TP53 в одиночных клонах ГСК может превратить «проигравших» в «победителей», что согласуется с наблюдаемым рецидивом соматических мутаций TP53 при КГ и миелоидных новообразованиях, возникающих при SDS и нарушениях биологии теломер.
Революция в области анализа отдельных клеток выявила гетерогенность популяций определенных типов клеток, и все чаще признается, что клональная экспансия отдельных клеток лежит в основе ряда заболеваний, помимо рака, включая сосудистые патологии, цирроз и нейродегенерацию (Brunner et al., 2019; Chappell et al., 2016; Dobnikar et al., 2018; Jacobsen et al., 2017; Misra et al., 2018; Sheikh et al., 2015; Tay et al., 2017). Например, множественные предшественники гладкомышечных клеток (ГМК) дают начало нормальной артериальной стенке во время развития, но несколько избранных предшественников внутри стенки участвуют в формировании атеросклеротических бляшек (Chappell et al., 2016; Greif et al., 2012; Jacobsen et al.., 2017; Misra et al., 2018). Кроме того, при возрастном клональном кроветворении с неопределенным потенциалом (от англ. clonal haematopoiesis of indeterminate potential или сокращенно – CHIP) стволовые клетки, несущие соматические мутации, дают доминантные варианты лейкоцитов, а CHIP ассоциируется с повышенным риском серьезных заболеваний, связанных с атеросклерозом, инфарктом миокарда и ишемическим инсультом (Jaiswal S. and Libby, 2020; Jaswal S. et al., 2017).
Клональный гемопоэз неопределенного потенциала (CHIP) уже некоторое время привлекает внимание медицинского сообщества. Как утверждают A. M. Cacic, F. I. Schulz, U. Germing et al. (2023), этот феномен, обнаруженный около десяти лет назад, связывает возрастные изменения кроветворения не только с более поздним развитием гематологических злокачественных новообразований, но и с повышенным риском ранних сердечно-сосудистых заболеваний и некоторых других заболеваний. CHIP выявляется в крови и характеризуется клонально расширенными соматическими мутациями в генах, связанных с раком, предрасполагающих к развитию гематологических новообразований, таких как миелодиспластический синдром (МДС) и острый миелолейкоз (ОМЛ). Связанные с CHIP мутации часто затрагивают гены восстановления повреждений ДНК и нередко отмечаются после предшествующей цитотоксической терапии рака. Генетическая предрасположенность, по-видимому, является фактором, способствующим этому. Стало неожиданностью, что CHIP значительно повышает риск инфаркта миокарда и инсульта, а также способствует развитию сердечной недостаточности и легочной гипертензии.
Недавние исследования на людях указывают на подобное прогрессирующее накопление мутантных соматических клонов в эпителиальных клетках различных органов при нормальном старении (Martincorena et al., 2018; Martincorena et al., 2015; Moore et al., 2020; Yoshida et al., 2020). Исследования в основном были сосредоточены на автономных клеточных механизмах, лежащих в основе клональной экспансии, а неклеточная автономная регуляция недостаточно хорошо изучена, особенно при старении и возрастзависимых болезнях цивилизации. Примечательно, что недавнее неоднозначное биоинформатическое исследование ставит под сомнение точку зрения о том, что увеличение соматических мутаций с возрастом лежит в основе повышенной заболеваемости раком в более позднем возрасте, и вместо этого утверждает, что снижение иммунной системы имеет первостепенное значение (Palmer et al., 2018).
Y. Ran, Z. Gu, Y. Lyu et al. (2022) исследовали феномен неклеточной автономной регуляции клональности при старении, определяя роль старых гемопоэтических клеток в модулировании рекрутирования и клональной экспансии клеток-предшественников (КП) гладкомышечных клеток (ГМК) в атеросклеротической бляшке. Они показали, что воспалительные клетки, особенно макрофаги, и КП ГМК играют ключевую роль в атерогенезе. Редкие предшественники ГМК из средней оболочки вносят свой вклад в клетки, которые заселяют зарождающуюся бляшку и клонально расширяются, приводя к ∼30—70% клеточности развитой бляшки с клетками, формирующими защитный фиброзный колпачок или переходящими в дестабилизирующие судьбы бляшки (Basatemur et al., 2019; Chappell et al., 2016; Jacobsen et al., 2017; Misra et al., 2018; Shankman et al., 2015; Wirka et al., 2019). Интегрины представляют собой гетеродимерные белки, которые связывают внеклеточные и внутриклеточные компартменты, а трансплантат костного мозга (BM), лишенный гена, кодирующего интегрин β3 (Itgb3), у мышей с атеропронами, индуцирует множественные предшественники SMC для проникновения в бляшку и клонального расширения, усугубляя бремя болезни (Misra et al., 2018; Schneider et al., 2007). На фоне Apoe (-/-) макрофаги, происходящие из Itgb3 (-/-) КМ, имеют повышенные уровни цитокина фактора некроза опухоли (TNF) -α (Schneider et al., 2007). Уровни TNFα в плазме повышены у старых мышей и людей, а высокие уровни TNFα у пожилых людей коррелируют с атеросклерозом (Bauernfeind et al., 2016; Bruunsgaard et al., 2000). Важно отметить, что трансплантация КМ (ТКМ) от старых мышей молодым мышам усугубляет атеросклероз (Du et al., 2016), но основные механизмы и эффекты на рекрутирование и клональность SMC не определены.
При CHIP клонально расширенные гемопоэтические стволовые клетки обычно содержат соматические мутации в эпигенетических регуляторах, таких как Ten Eleven Translocation (TET) – 2 (Jaiswal and Libby, 2020; Jaiswal et al., 2017). На нулевом фоне рецептора липопротеинов низкой плотности (Ldlr) КМ с дефицитом TET2 предрасполагает мышей к развитию атеросклероза, вызванного усиленной западной диетой, а макрофаги, выделенные от мышей TET2 (-/-), экспрессируют повышенные уровни цитокинов, включая TNFα. (Fuster et al., 2017; Jaiswal et al., 2017). Макрофаги, привлеченные к ранним атеросклеротическим бляшкам, локально пролиферируют во время прогрессирования заболевания (Robbins et al., 2013), а у мышей с атеропрозом инициация БВ у пожилых людей по сравнению с молодыми увеличивает накопление макрофагов в аорте (Du et al., 2016). Важно отметить, что в контексте CHIP у людей, или у старых мышей, или мышей с дефицитом миелоидных клеток TET2 клональность миелоидных клеток в самих атеросклеротических бляшках неуловима. Подобно старению, влияние дефицита КМ TET2 на клональность ГМК в бляшке не определено.
Показано, что возраст КM является ключевым фактором, определяющим клональность клеток, происходящих из ГМК, в атеросклеротической бляшке: состарившийся неклеточный КМ автономно индуцирует рекрутирование и экспансию множества предшественников ГМК. Механически сниженные уровни TET2 в старых моноцитах/макрофагах эпигенетически снижают экспрессию гена Itgb3, что усиливает передачу сигналов TNFα-TNF рецептора 1 (TNFR1). Таким образом, возникает поликлональность линии ГМК и более выраженный атеросклероз (Ran Y., Gu Z., Lyu Y. et al., 2022).
Но вопрос о том, что же является мотором и главным двигателем патогенеза и прогрессирования болезней цивилизации (БЦ) и постоянно поддерживает эту «неустойчивость генома» высокоспециализированных клеток организма при наличии или отсутствии генетического дефекта в их геноме и почему приводит к закономерному исходу болезни, очень долго оставался открытым. На основании почти 35-летнего опыта применения различных типов клеточной терапии у человека мы пришли к выводу, что таким молекулярно-биологическим двигателем является клональный гематопоэз, обусловленный геномным и постгеномным повреждением аутологичных гемопоэтических стволовых клеток (ГСК) и гемопоэтических клеток-предшественников (ГКП) костного мозга. ГСК и ГПК – главные системообразующие и главные регуляторные клеточные системы крови и родоначальники всех иммунокомпетентных клеток иммунной системы организма млекопитающих и человека! Именно ГСК и ГПК формируют системообразующие иммунноспецифические (иммунотолерантные или иммуноагрессивные) реакции организма человека и животных на клетки с неустойчивым геномом и определяют их судьбу. Это показано новыми знаниями о том, что ГСК и ГКП, с одной стороны, являются самыми медленными клетками в организме (их клеточный цикл от 22 до 50 недель или почти 360 дней) и формируют все иммунные реакции и иммунную память в организме (Seiweke М. et al., 2021). С другой стороны, кроме функций репродукции клеток крови и иммунокомпетентных клеток крови, а также формирования иммунных реакций, они являются главными регуляторными и управляющими системами организма всех млекопитающих. Под их управлением и в подсистемах к ним находятся все 230 известных типов клеток организма человека (Alberts B., Johnson A., Lewis J. еt al., 2007). Изменение геномной и протеомной структуры ГСК и ГПК, как самых длительно живущих клеток в организме, происходит в результате накопления в них драматических дополнительных соматических мутаций (ДСМ). В результате формируются и постепенно расширяются патологические клоны ГСК и ГКП и их потомки начинают доминировать в гемопоэзе и циркуляторном русле (Jaiswal S., Ebert B. L., 2019; Mitchell E., Chapman M. S., Williams N. et al., 2022).
Как отмечает в своем научном обзоре S. Jaiswal (2020), революция в области анализа отдельных клеток выявила гетерогенность популяций определенных типов клеток, и сегодня признается, что клональная экспансия отдельных клеток лежит в основе этиопатогенеза ряда таких заболеваний, как рак, сосудистые заболевания, цирроз и нейродегенерации (Brunner et al., 2019; Chappell et al., 2016; Dobnikar et al., 2018; Jacobsen et al., 2017; Misra et al., 2018; Sheikh et al., 2015; Tay et al., 2017). Парадокс заключается в том, что, например, множественные предшественники гладкомышечных клеток дают начало нормальной артериальной стенке во время развития, но несколько избранных предшественников внутри стенки участвуют в формировании атеросклеротических бляшек (Chappell et al., 2016; Greif et al., 2012; Jacobsen et al. 2017; Misra et al., 2018). Кроме того, при возрастном клональном кроветворении стволовые клетки, несущие соматические мутации, дают доминантные варианты лейкоцитов, а появление патоспецифических клонов связано с повышенным риском серьезных заболеваний, асоциированных с атеросклерозом, инфарктом миокарда и ишемическим инсультом (Jaiswal S. & Lybbi, 2020; Jaiswal S. et al., 2017). Недавние исследования на людях указывают на подобное прогрессирующее накопление мутантных соматических клонов в эпителиальных клетках различных органов, что характерно и для нормального старения (Martincorena et al., 2018; Martincorena et al., 2015; Moore et al., 2020; Yoshida et al., 2020). Показано, что ДСМ со временем накапливаются во всех клетках (Welch J. S., Ley T. J., Link D. C. et al., 2012; Alexandrov L. B., Jones P. H., Wedge D. C. et al., 2015; Martincorena I., Campbell P. J., 2015; Blokzijl F., de Ligt J., Jager M. et al., 2016; Hoang M. L., Kinde I., Tomasetti C. et al., 2016). Эти ДСМ чаще всего представляют собой замены оснований (известные как однонуклеотидные варианты), небольшие вставки, или делеции (indels), или изменения числа копий больших хромосомных областей (известные как структурные варианты). По оценкам, ГСК приобретают примерно 20 соматических мутаций в год во всем геноме (Lee-Six H., Øbro N. F., Shepherd M. S. et al., 2018; Osorio F. G., Rosendahl Huber A., Oka R. et al., 2018) и примерно 0,1 мутации в год в экзонах, кодирующих белок (Welch J. S., Ley T. J., Link D. C. et al., 2012), подавляющее большинство из которых являются однонуклеотидными вариантами. В костном мозге только долгоживущие ГСК обладают способностью к самообновлению на протяжении всей жизни организма (Reya T., Morrison S. J., Clarke M. F., Weissman I. L., 2001). Поэтому в большинстве случаев только мутации, возникающие в ГСК, сохраняются на протяжении всей жизни человека. Учитывая, что на человека приходится от ∼50 000 до ∼200 000 ГСК, считается, что к 70 годам у людей насчитывается от 350 000 до 1 400 000 кодирующих мутаций в пуле ГСК. Если хотя бы одна из этих мутаций способна обеспечить селективное преимущество в отношении ГСК, в котором она возникает, клональная экспансия в крови должна быть обычным явлением при старении (Jaiswal S., Ebert B. L., 2019). Действительно, это явление, называемое клональным гематопоэзом, тесно связано со старением и показано в нескольких исследованиях лиц, не отобранных для исследования гематологических нарушений (Xie M., Lu C., Wang J. et al., 2014; Genovese G., Kähler A. K., Handsaker R. E. et al., 2014; Jaiswal S., Fontanillas P., Flannick J. et al., 2014; McKerrell T., Park N., Moreno T. et al., 2015; Buscarlet M., Provost S., Zada Y. F. et al., 2017; Acuna-Hidalgo R., Sengul H., Steehouwer M. et al., 2017; Coombs C. C., Zehir A., Devlin S. M. et al., 2017).
По мнению S. Jaiswal (2020), в большинстве исследований мутации, используемые для определения клонального кроветворения, аналогичны мутациям, обнаруживаемым при гематологическом раке (Jaiswal S., Fontanillas P., Flannick J. et al., 2014; Coombs C. C., Zehir A., Devlin S. M. et al., 2017). Наиболее часто мутирующие гены в клональном кроветворении включают DNMT3A, TET2, ASXL1, JAK2, TP53 и SF3B1, которые также часто мутируют при остром миелоидном лейкозе (Ley T. J., Miller C., Ding L. et al., 2013; Lindsley R. C., Mar B.G., Mazzola E. et al., 2015), миелодиспластическом синдроме (МДС) (Bejar R., Stevenson K., Abdel-Wahab O. et al., 2011; Papaemmanuil E., Gerstung M., Malcovati L. et al., 2013) и миелопролиферативных новообразованиях (МПН) (Nangalia J., Massie C. E., Baxter E. J. et al., 2013). Следовательно, неудивительно, что у людей с клональным гематопоэзом эти виды рака развиваются с большей скоростью, чем у людей без мутаций (Genovese G., Kähler A. K., Handsaker R. E. et al., 2014; Abelson S., Collord G., Ng S. W. K. et al., 2018; Jaiswal S., Fontanillas P., Flannick J. et al., 2014; Desai P., Mencia-Trinchant N., Savenkov O. et al., 2018; Bolton K. L., Ptashkin R. N., Gao T. et al., 2019). Однако мутации, вызывающие клональное кроветворение, также могут быть обнаружены в циркулирующих иммунных клетках, таких как гранулоциты, моноциты и лимфоциты. Это открытие повышает вероятность того, что клональное кроветворение может привести к измененным иммунным ответам, которые потенциально могут влиять на многие болезни старения (Mitchell E., Chapman M. S., Williams N. et al., 2022).
Клональное кроветворение относится к любому состоянию клональной экспансии в кроветворной системе. Рак крови, такой как хронический миелоидный лейкоз или МДС, является типичным примером клонального кроветворения. Однако те же самые мутации, обнаруженные при этих видах рака, также наблюдаются у значительной части здорового пожилого населения. Чтобы отличить наличие этих мутаций в незлокачественных условиях от злокачественного клонального кроветворения, был введен термин «клональный гематопоэз с неопределенным потенциалом» (CHIP) (Steensma D. P., Bejar R., Jaiswal S. et al., 2015). CHIP определяется наличием связанной с раком соматической мутации в крови или костном мозге у лиц без известных гематологических раковых заболеваний или других клональных состояний, таких как моноклональная гаммапатия.

