



Виталий Давидов
Боль в пояснице
Для проведения питательных веществ через мембранный механизм замыкательной пластинки затрачивается около 2 часов (в среднем на 0,7 мм толщины пластинки) сквозь мембрану и еще около 4-5 часов транспортировки в центр диска. В среднем, в здоровом диске на доставку питательных веществ от границы капиллярного русла до центра диска затрачивается около 7-8 часов. И это при условии адекватной пропускной способности замыкательных пластин и полноценной работы капиллярной сети позвонков. Поэтому, чем выше диск (например, L4-5), тем больше времени понадобиться для прокачки питательных веществ с периферии. Выведение продуктов обмена идет по тому же пути диффузии, но только в обратном направлении. Поэтому популяция клеток центральной части диска всегда будет более уязвима в отношении питания и выведения продуктов обмена. Очень часто это физически невозможно, если нарушена диффузия через замыкательные пластины или нарушен капиллярный кровоток. Склерозирование замыкательных пластин может развиться после травмы или в результате длительного воздействия статических нагрузок. Нарушение пропускной способности капилляров, как правило, вызвано их спазмом в результате воздействия никотина (курение), вибрации (большие автомобили), снижением насосной функции мышц (длительное сидение) и сосудистыми заболеваниями (атеросклероз, диабет и др.).
В любом случае, доставка питательных веществ снижается настолько, что клетки не в состоянии поддерживать свой нормальный жизненный цикл. При этом также нарушено выведение продуктов обмена и идет процесс «самоотравления» клеток.
Поэтому естественная ходьба и кратковременные компрессионные нагрузки средней интенсивности улучшают качество диффузии и выступают анаболическим фактором, способствуя выработке клетками пульпозного комплекса структурных белков и коллагена II типа. Длительное нахождение в положении лежа, сидя или длительные компрессионные нагрузки даже низкой интенсивности выступают катаболическим фактором, приводя к ухудшению диффузии и снижению обменных процессов.
Анаболическое «окно» полноценного обмена в диске лежит в пределах 0,3-1,3 МРа (МПа; мегапаскаль). Для ориентировки: положение лежа – 0,1 МРа, компрессионная нагрузка может приводить к повышению давления до 3,0 МРа. Вся нагрузка ниже или выше значений 0,3-1,3 МРа является катаболической.
Осмотическое (гидростатическое) давление в диске лежит в диапазоне 420-450 mOsm, при этом оптимальное давление для выработки протеогликанов и экстраклеточного матрикса лежит в диапазоне 400 -500 mOsm (изоосмотическое и гиперосмотическое).
Кратковременный катаболизм тканей диска особой угрозы не представляет. Но, если речь идет о годах «катаболического стажа», тогда существует повышенный риск развития дегенеративного заболевания диска – межпозвонкового остеохондроза с последующим развитием в остеохондроз позвоночника. [Здесь нет никакой тавтологии. Это похожие по названию, но разные по клиническому проявлению «заболевания»].
При осмотическом (гидростатическом) давлении 0,3 МРа клетками синтезируется на 20% больше протеогликанов, чем при давлении 0,1 МРа (лежа). Высокая, но даже кратковременная компрессионная нагрузка выступает катаболическим фактором: замедляет, прекращает синтез структурных белков и вызывает их распад.
Весомым и, зачастую необоснованно игнорируемым фактором нарушения обменных процессов, является длительное сидение. Диск может выдержать кратковременную компрессионную нагрузку до 250 кг на см квадратный без последствий (до 9000N), но он бессилен против обычного длительного сидения изо дня в день из года в год. В здоровом диске снижение тургора наблюдается уже через 45 минут сидения. В стадии дистрофии диск «проседает» и «распластывается» уже через 20.
Отсюда возникает первая проблема – нарушение обменных процессов в диске при недостаточности диффузии.
Нарушение обменных процессов в диске при недостаточности диффузии
Обменные процессы в клетках зависят от ряда факторов: генетических, прилагаемой физической нагрузки, состояния периферических сосудов и пропускной способности замыкательных пластин.
В процессе жизнедеятельности диски постоянно подвергаются нагрузке, что приводит к частичному разрушению элементов матрикса и необходимости продукции новых аггрекановых комплексов на замену поврежденным.
В здоровом диске постоянно происходит смена анаболической и катаболической фаз. Клетки диска вырабатывают специальные ферменты (энзимы), которые разрушают и уничтожают поврежденные и отработавшие срок структурные элементы. Аггреканазы 1,2 (ADAMTS 4,5) «разрушают» и «растворяют» отработавший дефектный аггрекановый комплекс; матриксные металлопротеиназы (ММР 1,3 и др.) растворяют части поврежденного матрикса. Действие матриксных металлопротеиназ сбалансировано действием фермента-антагониста – тканевого ингибитора матриксных металлопротеиназ (TIMP 1 и 2), который блокирует (подавляет) действие основного «растворителя» и препятствует избыточному разрушению матрикса. TIMP 3 блокирует действие аггреканаз. Смена катаболической и анаболической фазы позволяет в текущем режиме устранять повреждения и проводить «чистку» внутренней среды диска. Т.е. постоянно происходит так называемое гомеостатическое ремоделирование пульпозного ядра.
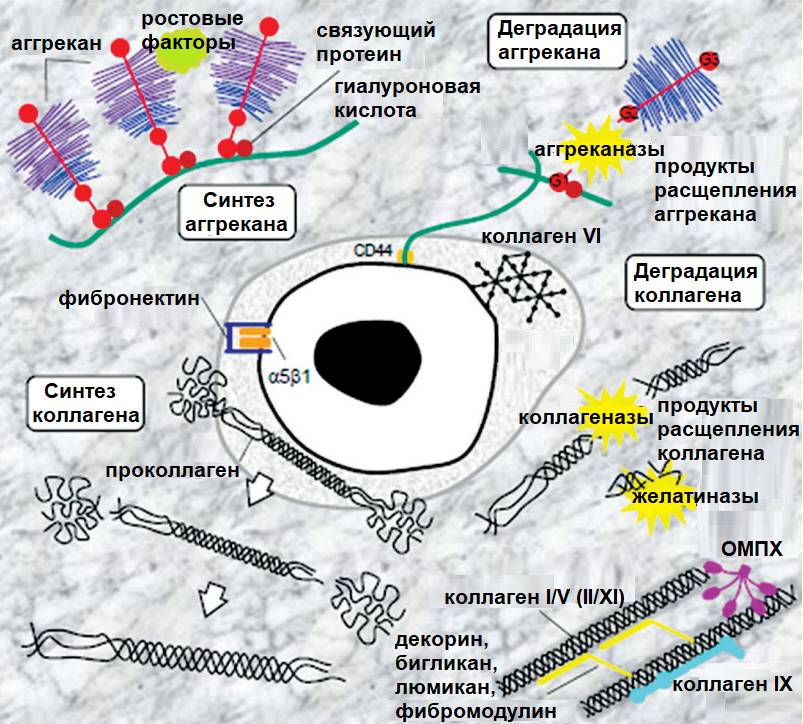
Хондроцитоподобная клетка и белки внеклеточного матрикса (Bendtsen et al., 2016)
Оптимальной средой для клеточного анаболизма является нейтральная среда. Колебания pH в диске лежат в диапазоне от 7,5 до 5,7. pH здорового диска – 7,1. Максимальный синтез структурных белков при pH 7,0. Клетки пульпозного ядра адаптированы работать в кислой и умеренно ишемической среде. Напряжение кислорода в пульпозном ядре может колебаться в диапазоне от 10% (нормоксия) до 0,5% (критическая гипоксия). Оптимальное для клеток – 1,5-5% (функциональная гипоксия). На периферии фиброзного кольца напряжение кислорода достигает 19%; центр пульпозного комплекса – 0,65%.
Снижение pH до 6,8 – прекращается синтез структурных белков пульпозного комплекса. Снижение pH до 6,5 ведет к дегенерации диска. Рабочий глюкозный диапазон в пределах от 0,8 до 5 mmol/L. Падение уровня глюкозы ниже 0.5 mmol/L больше чем на три дня и снижение pH ниже 6.4 приводит к смерти клеток.
Утрируя: смерть клеток пульпозного ядра – это смерть всего диска.
В природе существует запрограммированная смерть клеток (апоптоз) и регулируемая смерть клеток (пироптоз, некроптоз, ферроптоз).
Смерть клетки вызывается двумя механизмами: внутренним и внешним.
Внутренний путь смерти клетки вызван оксидативным стрессом, стрессом эндоплазматического ретикулума, повреждением ДНК или повреждением митохондрий. Итогом является активация цитозольных белков, отвечающих за апоптоз, с последующей деструкцией ядерных белков и цитоскелета, синтеза «рецепторов смерти», хемокинов (приманок) с целью привлечения и активации клеток, выполняющих фагоцитирующие функции.
Внешний механизм смерти клетки связан с активацией «рецепторов смерти» с помощью «лигандов смерти», синтезируемых клетками иммунной системы.
Для удобства восприятия выделяют три последовательные фазы дегенерации диска: 1) катепсинозависимая дегенерация (макрофагонезависимая), 2) воспалительная макрофагозависимая и 3) репаративная макрофагозависимая фаза. При травме диска дегенерация начинается сразу с воспалительной макрофагозависимой.
К пусковым причинам дегенерации диска относят: ишемию, ацидоз (критическое накопление лактата), прямую травму, оксидативный стресс, стресс эндоплазматического ретикулума, накопление продуктов гликации, метаболические нарушения на фоне генетической предрасположенности.
Ишемия и ацидоз. Катепсинозависимая дегенерация – остеохондроз бухгалтеров, водителей и прочих сидячих профессий. Метаболическое поражение диска.
Самый частый вопрос: «Ничего тяжелого не поднимаю, больших физических нагрузок нет. Почему начала болеть спина и почему появился остеохондроз?». Ответ прост: нарушение обменных процессов в диске, связанное с длительным «стажем» сидения.
Пусковым фактором выступает хроническая критическая ишемия диска вследствие длительного кислородного голодания и «закисания» диска. Снижение напряжения кислорода до 1 – 0,5 % и параллельное снижение pH вследствие накопления лактата приводит к ускоренному старению и смерти клеток пульпозного ядра.
Само по себе сидение вызывает очень низкую компрессионную нагрузку, которая не обеспечивает достаточный «помповый» механизм откачки метаболитов.
Для поддержания нормальной жизнедеятельности клеткам нужна энергия. Она производится двумя способами: оксидативное (окислительное) фосфорилирование (с участием кислорода) и гликолиз (без участия кислорода). Другими словами, энергия добывается или «горением», или «брожением». В обильно кровоснабжаемых тканях клетки предпочитают синтезировать энергию с участием кислорода (акцептор электронов), но подобная практика сопряжена с повышенным синтезом побочных продуктов – активных форм кислорода (АФК; Reactive oxygen species, ROS) и оксидативным стрессом, что может привести к повреждению митохондрий и гибели клеток. Обильное кровообращение (соответственно, обмен) позволяет в ускоренном режиме утилизировать побочные метаболиты и/или избавляться от погибших клеток. Межпозвонковый диск является самым крупным бессосудистым органом в теле человека, что жестко детерминирует его метаболизм. Оксидативное фосфорилирование как базовое не подходит в принципе, но кислород и «побочные» метаболиты митохондриального дыхания нужны клеткам как вторичные мессенджеры для запуска и контроля важных сигнальных путей, связанных с жизнедеятельностью клеток. Поэтому 75% энергии в клетке синтезируется благодаря анаэробному гликолизу, 25% – аэробному пути обмена.
За оксидативное энергообеспечение отвечает коактиватор-1α (PGC-1α), за гликолитическое – транскрипционный фактор под названием Фактор, индуцируемый гипоксией (Hypoxia-inducible factor 1-alpha; HIF).
Сам Фактор двухкомпонентный: субъединицы 1 альфа, 2 альфа и 3 альфа и субъединица 1 бета. Соединяясь воедино в ядре клетки, HIF инициирует транскрипцию различных генов в зависимости от «комбинации пары»: 1) HIF-1α + HIF-β; 2) HIF-2α + HIF-β; 3) HIF-3α + HIF-β.
Изоформа HIF-1α является кислородным сенсором клетки и при падении напряжения кислорода ниже 5% связывается с HIF-β, формируют активный HIF, который активирует транскрипционные факторы/гены, чувствительные к гипоксии: NOS2(Индуцибельная синтаза оксида азота), VEGF (Фактор роста эндотелия сосудов), EPO (эритропоэтин), GLUT1 (глюкозный транспортер), SOX9 (регуляция экспрессии гена коллагена II типа в хондроцитах) , IGF2 (инсулиноподобный фактор роста-2), COL2A1 (коллаген II типа) и другие, отвечающие за поддержание жизнедеятельности клетки «на пределе». Данный фактор позволяет работать клеткам в оптимальном гипоксическом диапазоне между 2-5% парциального напряжения кислорода, при котором синтезируется больше всего аггрекана и коллагена II типа. HIF-1α – это мастер-регулятор гликолитического метаболизма клеток при ишемии, который регулирует уровень внутриклеточного (цитозольного) лактата. Это один из факторов, отвечающих за пролиферацию (деление) клеток, дифференцировку незрелых стволовых клеток, поддержание их метаболической активности, синтез коллагена II типа, регуляцию синтеза экстраклеточного матрикса, регуляцию ангиогенеза, регуляцию аутофагии, апоптоз и минерализацию замыкательной пластинки через активацию адаптивных генов.
Изоформа HIF-2α – является катаболическим регулятором и отвечает за активацию генов синтеза матрикс деградирующих энзимов (протеиназ) и провоспалительных медиаторов. Индуцируется фактором некроза опухоли- альфа (TNF-α) и через активацию ядерного фактора κB (NF-κB) запускает синтез ММР-3 и ММР-13, стимулирует оссификацию замыкательной пластинки, блокирует аутофагию в клетках, стимулирует ангиогенез (рост сосудов).
Изоформа HIF–3α является регулятором изоформ HIF-1α и HIF-2α, блокируя их деятельность.
При напряжении кислорода в пульпозном ядре порядка 1-5%, «обобщенный» HIF активирует как сигнальные пути, запускающие анаболические процессы в клетках через активацию генов, отвечающих за синтез аггрекана и коллагена II типа, так и сигнальные пути, запускающие катаболические процессы через активацию генов, отвечающих за синтез интерлейкина-1бета (IL-1β) , фактора некроза опухоли – альфа (TNF-α) и т.д. Другими словами, фактор отвечает за баланс анаболических и катаболических процессов (гомеостатическое ремоделирование матрикса пульпозного ядра при гипоксии) и работает по принципу стрелочного перевода на железной дороге.
При падении напряжения кислорода ниже 1% (критическая гипоксия) фактор HIF активирует только катаболические пути и, по сути, являясь химическим детонатором воспалительной реакции в диске, итогом которой будет смена фенотипа («старение» клеток) или апоптоз клеток (смерть) с последующими дегенеративными процессами в матриксе диска. Длительная пороговая гипоксия (0,5 – 1%) вызывает «перебор» по синтезу HIF фактора, который в том числе активирует гены, ответственные за синтез фактора роста эндотелия сосудов (VEGF). Это компенсаторная реакция, направленная на восстановление дыхательного гомеостаза пульпозного комплекса за счет стимула врастания капилляров внутрь диска.
Нарушение синтеза HIF может привести к смерти клеток из-за обратного процесса – выраженного повышения парциального напряжения кислорода на фоне повышения уровня глюкозы (нарушена взаимная балансировка по оси «HIF-PGC-1α»). При таком анаболическом раскладе резко повышается производство энергии митохондриями по оксидативному (!) типу с быстрым накоплением перекисей и дальнейшим оксидативным стрессом. Клетки пульпозного ядра предпочитают работать в умеренно ишемической среде за счет гликолитического окисления углеводов и жиров («брожение»). Это снижает количество утечек электронов из дыхательной цепи (в норме только 0,2 – 2%) и уменьшает объем синтезируемых активных форм кислорода (АФК), предотвращая оксидативный стресс.
Другой процесс. Перебор по синтезу HIF-1а приведет к блокировке PGC-1α, который отвечает за митохондриальный генез (фрагментация митохондрий и их «склеивание» с увеличением размера и дыхательного потенциала за счет увеличения крист). PGC-1α отвечает за активацию генов, ответственных за лизис («растворение») поврежденных митохондрий, накопление которых также вызывает оксидативный стресс за счет высокой утечки АФК. Митохондрии лучше представить как «клетка в клетке». Повреждение большого количества митохондрий для клетки крайне нежелательно. Полная блокировка работы PGC-1α приведет к накоплению митохондриального мусора и спровоцирует гибель клетки из-за переизбытка вовремя не утилизированных «фрагментов» и активных форм кислорода (АФК).
АФК являются разновидностью нестабильных и высокоактивных молекул, включая
гидроксильный радикал (ОН-); супероксид-анион (O2–); перекись водорода (H2O2); оксид азота (NO) и гипохлорит-ион (OC1-). Они образуются как побочные продукты клеточного аэробного метаболизма и являются важными внутриклеточными сигнальными молекулами, участвующими в регуляции различных физиологических процессов в клетках. Но это если речь идет о нормальном (допустимом) их количестве. Под оксидативным стрессом подразумевают комплексное повреждение органелл клетки и структурных биомолекул. При избыточном производстве и накоплении, активные формы кислорода встраиваются в молекулы и приводят к повреждению внутриклеточных протеинов, нуклеиновых кислот, мембранных липидов и углеводов. Чисто механически, АФК просто химически «ломают» молекулы, превращая их в клеточный «мусор». Или что еще хуже – изменяют их конформационные свойства за счет образования новых патологических структурных молекул. Наиболее подвержены повреждению серосодержащие аминокислоты цистеин и метионин, что приводит к банальному распаду белков. Оксидативный стресс может вызывать агрегацию (соединение) протеинов и превращать их в цитозольный «патоген» (аутоантиген). Например, окисленные белковые продукты (advanced oxidative protein products (AOPPs) могут активировать сигнальный путь MAPK и запускать апоптоз. При оксидативном повреждении липидов образуются активные формы карбонила (reactive carbonil species) – которые повреждают как сами липиды (мембраны/оболочки), так и протеины, нуклеиновые кислоты. Активные формы карбонила способствуют образованию конечных продуктов гликирования (Advanced glycation end products (AGEs) – «склеившиеся» белки или липиды в результате воздействия сахаров. «Засахаренные» молекулы крайне опасны для организма. Они приводят к митохондриальной дисфункции, нарушая работу митохондриальных пор, что вызывает снижение уровня синтеза белка Bcl-2 (антиапоптического белка), контролирующего проницаемость мембран, и активацию Bax – белка, увеличивающего проницаемость митохондриальной мембраны и стимулирующего запуск апоптоза.
АФК ломают молекулы ДНК, что вынуждает клетки «стопорить» деление вследствие нежелания воспроизводить свои «дефектные» копии. Также активные формы кислорода повреждают сами митохондрии (разрыв оболочки), что приводит к нарушению их работы и запускает каскад цепных реакций, приводящих к старению клетки или ее смерти. Здоровые клетки способны препятствовать накоплению АФК через активацию редокс («очистных») систем (синтез супероксид дисмутаз, каталаз и глутатиона; Nf2 зависимые гены). Нарушение оксидативно-восстановительного статуса при дегенерации МПД с одной стороны проявляется в виде увеличения продукции АФК, с другой стороны – снижением синтеза и активности антиоксидантных веществ.
Поэтому поддержание выживаемости клеток при низком pH и низком напряжении кислорода зависит от способности клеток синтезировать и вовремя утилизировать фактор, индуцируемый гипоксией-1альфа (HIF-1α) и коактиватор-1альфа (PGC-1α).
HIF-1α напрямую влияет на метаболизм глюкозы: регулирует работу глюкозных транспортеров GLUT1, GLUT3 и GLUT9, ответственных за доставку глюкозы в клетку. Фактор поддерживает лактатный гомеостаз клеток и не позволяет им «перекисать», «стареть» и гибнуть. Также влияет на процесс митофагии (устранения поврежденных митохондрий). HIF-1α блокирует Fas-рецептор («рецептор смерти»), тем самым защищая клетки от Fas-лиганд опосредованного апоптоза при атаке иммунными клетками. Активность фактора связана с циркадными ритмами и напряжением кислорода. Высокое напряжение кислорода (6-10% при врастании капилляров) деактивирует субъединицу HIF-1α и не позволяет ей смещаться в ядро клетки, связываться со своим «напарником» HIF-1β, формировать активную форму и активировать транскрипцию нужных генов. HIF циркадно зависимый от протеинов «циркадных часов». Чтобы фактор синтезировался, нужен полноценный сон. У людей, работающих на ночных сменах, боль в спине встречается чаще.
Сигнальные пути HIF и NF-κB пересекаются, что объясняет связь ишемии с дальнейшим воспалением. Одной из главных задач фактора является синтез белков-насосов, откачивающих лактат за пределы клетки. Для удаления излишка, ранее «откачанный» в межклеточный матрикс лактат должен быть выведен из диска при помощи диффузии (порядка 8 часов от центра пульпозного ядра). В норме при полноценной диффузии все продукты клеточного метаболизма успешно выводятся из диска и утилизируются. Качество диффузии зависит от регулярной длительной ходьбы и 8-часового сна (ночная «очистка» диска). «Хроническое» сидение как раз и нарушает «помповый» механизм откачки лактата. При сидении нагрузка на диск 0,2 МРа, поэтому она и относиться к катаболической.
Параллельное повышение внеклеточного невыведенного лактата является ключевым механизмом, усиливающим дегенерацию диска при межпозвонковом остеохондрозе: на мембранах клеток пульпозного ядра и замыкательной пластинки находятся рецепторы, чувствительные к закислению (Acid sensing ion channel; ASIC1 и ASIC3). Это универсальные вольтаж – независимые ионные каналы, которые выполняют множество функций в организме. При умеренном pH 7,0 рецепторы «заблокированы». По мере усиления ацидоза (закисания пульпозного комплекса) рецепторы активируются протонами водорода Н+, которые перекачиваются через мембраны митохондрий и накапливаются при синтезе АТФ. При утечке через поврежденные мембраны протоны в избытке появляются в цитозоле клетки. Затем выбрасываются клетками в межклеточное пространство. Высокие уровни лактата усиливают чувствительность ASIC к протонам водорода.
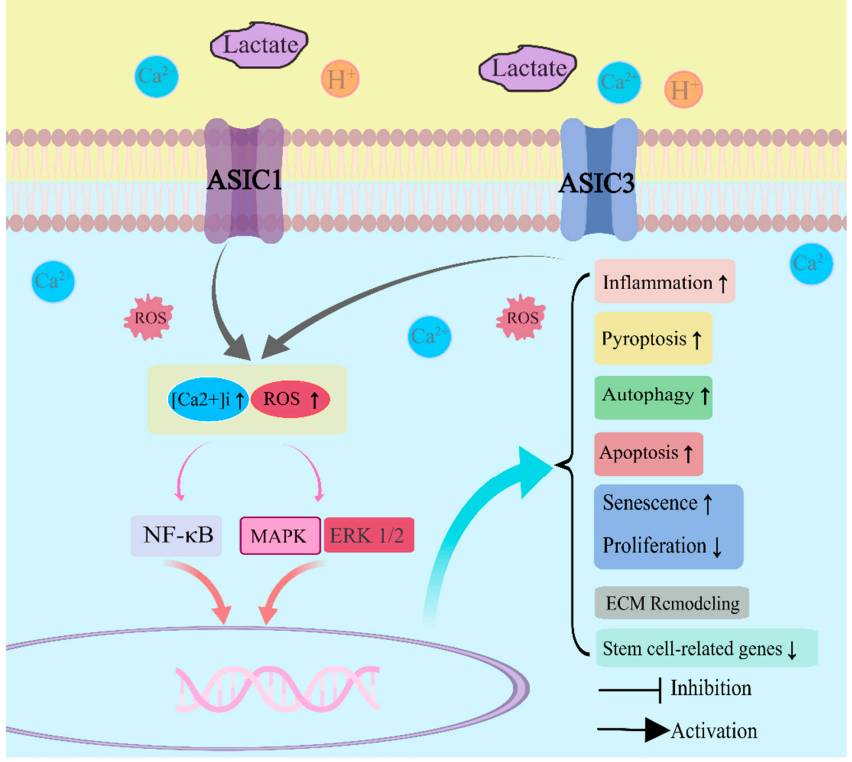
Гиперактивация кислотночувствительных каналов вызывает воспаление, увеличивает пироптоз, апоптоз, повышает аутофагию, ускоряет старение клеток, снижает их пролиферацию, влияет на ремоделирование матрикса, подавляет транскрипцию генов стволовых клеток (Luo et al., 2023).
Активация ASIC1 в хондроцитоподобных клетках приводит к резкому повышению внутриклеточного кальция Ca2+, что вызывает митохондриальную дисфункцию и эндоплазматический стресс, итогом которого будет оксидативное стресс-индуцированное старение клеток, апоптоз или пироптоз вследствие активация сигнального пути ядерного фактора κB (NF-κB), увеличение синтеза провоспалительных цитокинов, аггреканаз и металлопротеиназ. Рецептор ASIC3 в отличие от других типов кислотночувствительных рецепторов работает с некоторой задержкой закрытия, которая увеличивается по мере закисания ткани. Чем более кислая среда, тем дольше рецептор открыт и больше пропустит кальция в клетку.
Эта же судьба и хондроцитов замыкательной пластинки. ASIC3 работает в паре с фактором, индуцируемым гипоксией-1α (HIF-1α), и регулирует адаптационные возможности клеток к гиперосмотической среде в условиях умеренного ацидоза. Выраженное скисание диска приводит к компенсаторному увеличению количества рецепторов ASIC3 на мембране, что увеличивает количество входящего Ca2+. Дальше дегенерация развивается по двум сценариям:
Первый. Постепенная декомпрессия диска вследствие снижения осмотического давления. Умеренное, но затяжное течение.
Аггрекан является основным протеогликаном, имеющим критическое значение для поддержания осмотического давления (оптимального тургора диска). Осмолярность экстраклеточного матрикса в течении дня колеблется в пределах от 430 до 496 mOsm единиц, что требует подстройки метаболических процессов под разное давление. Обменные процессы в клетках зависят от осмочувствительного транскрипционного фактора (TonEBR, tonicity-responsive enhancer binding protein), который регулирует работу: 1) аквапоринов (AQP -1 AQP – 5), рецепторов, пропускающих в клетку воду и кислород; и 2) осмо-, и механочувствительных ванилоидных рецепторов переходного потенциала TRPV4, которые регулируют синтез коллагенов. При низком осмотическом давлении TRPV4 активируют гены провоспалительных цитокинов IL-1β и IL-6.
Быстрое разрушения матрикса и аггрекана приводит к снижению осмотического давления и увеличению синтеза IL-1β и IL-6 резидентными клетками. Позвоночно-двигательный сегмент с «просевшим» диском начинает усиленно «ерзать». Каждое движение сопровождается увеличением линейного сдвига вышележащего позвонка над нижележащим с увеличением нагрузки на фиброзное кольцо. Механочувствительный рецептор TRPV4 является рецептором, специализирующемся на тракции (растяжении) и первым отреагирует на подобную нагрузку. Правда, отреагирует специфически через активацию NF-κB и усиленным синтезом провоспалительных цитокинов, что усугубляет разрушение диска через патологическое ремоделирование матрикса. Параллельно синтезируемые клетками катепсины K, D, L и сериновые протеазы, выбрасываемые в межклеточное пространство, не только стимулируют усиленную выработку соседними клетками матриксных металлопротеиназ (MMP), но и при этом блокируют действие их тканевого ингибитора (TIMP), таким образом, многократно усиливая катаболическое (разрушающее) действие металлопротеиназ в расщеплении матрикса. Поэтому такую дегенерацию еще обозначают как катепсиновая или «макрофагонезависимая дегенерация», т.е. без привлечения клеток иммунной системы при повреждении диска. Интерлейкин-6 (IL-6) «позаботится» о ремоделировании лимбуса и формировании остеофитов.
Второй сценарий (более агрессивный) развивается, если не предпринять меры по стабилизации сегмента и восстановления нормального pH.
Механический стресс приводит к быстрому накоплению АФК, последующему повреждению митохондрий и эндоплазматического ретикулума, что закономерно приводит к активации сигнального пути ядерного фактора κB (NF-κB), который активирует гены, отвечающие за сборку инфламмасом. Инфламмасомы (англ. inflammasome от англ. inflammation – воспаление) – самособирающийся пул цитозольных белков, отвечающих за активацию воспалительного ответа через синтез и секрецию провоспалительных цитокинов интерлейкина-1β (IL-1β) и интерлейкина-18 (IL-18). IL-18 в клетках пульпозного ядра стимулирует сигнальный путь каспазы-3/9, способствуя апоптозу клеток и деградации экстраклеточного матрикса. Вне зависимости от типа патологического сигнала, клетка начинает заранее готовиться к воспалению и синтезирует адаптерные белки с доменами для пирина и прокаспазы-1 (неактивная форма каспазы). Плюс начинает синтезировать запасы предшественников интерлейкинов-1/-6/-8/-12. Каспаза 1 отвечает за клеточный иммунитет и является активатором интерлейкина-1β – главного воспалительного цитокина. Некоторое время клетка буквально складирует «заготовки» белков, ответственных за воспаление. В фазу подготовки к сборке инфламмасомы входит «накопление» кальция Ca2+ в цитоплазме, митохондриальная дисфункция, «накопление» активных форм кислорода, постепенное разрушение органелл (лизосом). Если любой патологический сигнал имеет достаточную силу/концентрацию или длится достаточное время, происходит сборка инфламмасомы с активацией каспазы-1, которая, в свою очередь, неактивные формы интерлейкина-1β переводит в активные. Также каспаза-1 активирует «спящий» белок гасдермин D c разделением его на активные формы C и N. Фрагмент гасдермина N соединяется с мембранными белками клетки и образует большие поры («дыры»), через которые наружу выбрасываются калий (К+), цитокины и IL-1β с одновременным резким входом воды. Клетка разбухает и буквально взрывается как «бешеный огурец», выстреливая провоспалительные цитокины во все стороны. Критическое (ниже 6,5 pH) скисание пульпозного ядра или резкий вход кальция с одновременным выходом калия вызывает прямую активацию инфламмасомы NLRP3 без задержек. Активация соседних клеток интерлейкином-1β и 18 приводит к выраженному усилению синтеза матриксных металлопротеиназ и аггреканаз.
Круг замкнулся: пироптозная смерть одних клеток запускает цепную реакцию с последующим быстрым «схлопыванием» диска.
Особенно опасна гиперактивность ASIC1. Это рецептор со специализацией по хондроцитам замыкательной пластинки. Его повышенная «деятельность» приводит к гибели хондроцитов и появлению дефектов в пластинке и просачиванию жидкой фракции пульпозного комплекса в тело позвонка. Причем этот «коктейль» крайне токсичен в силу большого содержания провоспалительных цитокинов. Это одна из причин воспаления в субхондральной части кости (изменения по Модику 1).

ASIC1а/ASIC3– кислотночувствительные рецепторы; Low pH – низкий рН; SMAD – белок трансдукции внешнего сигнала от трансформирующего фактора роста-бета (TGF-β); NGF – фактор роста нервов; HIF-1α – фактор, индуцируемый гипоксией-альфа 1; ACAN – гены аггрекана; Col2a1- гены коллагена II типа; MAPK – митоген-активируемая протеинкиназа. ERK – экстраклеточный-сигнал регулируемая киназа; MMP – матриксные металлопротеиназы. Красные стрелки – активация, черные «тупики» – блокирование. (Guo et al., 2019).
Интерес представляет собой генетический механизм защиты тканей от самоуничтожения: когда клетка «понимает» что ее деятельность угрожает целостности ткани, то она «жертвует» своей жизнью ради сохранения популяции здоровых и полноценных клеток и переходит в режим контролируемой «самоликвидации» по сценарию, не угрожающему соседним клеткам. Но очень часто предъявляемые нагрузки настолько критичны, что смерть наступает не от апоптоза, а от пироптоза: стимулов «смерти» настолько много, что клетку буквально «разрывает» от внутренних деструктивных химических процессов.
При апоптозе клетка «схлопывается», «скукоживается» и сжимается. Умирает тихо, мирно и под контролем внутриклеточных генетически обусловленных механизмов, регулирующих запрограммированную смерть клеток. Нет выброса ядерных белков в межклеточное пространство. В норме апоптоз не сопровождается воспалительной реакцией соседних клеток.
Критическая проблема санации пульпозного комплекса – невозможность быстрого и качественного удаления мертвых клеток вследствие отсутствия кровоснабжения диска и иммуннопривилегированности пульпозного комплекса. Соответственно, у макрофагов (профессиональных «чистильщиков») нет прямого доступа к «трупам». В норме в васкуляризированных тканях утилизация апоптозных «трупов» осуществляется за счет фагоцитоза, который обеспечивает минимальный риск попадания клеточного содержимого в межклеточное пространство. Нормальный фагоцитоз может быть обеспечен только резидентными или циркулирующими макрофагами. Умирающая клетка на последнем этапе жизни «сигнализирует» макрофагам с помощью хемокинов о необходимости ее поглощения и утилизации. Макрофаг захватывает клетку, перемещает ее в лизосому (внутренний центр «переваривания»), таким образом, предотвращая случайный выброс внутриядерных белков в межклеточное пространство. Если апоптическая клетка долго не захватывается макрофагами, то она подвергается разрушению различными энзимами/протеазами, что приводит к выбросу ядерных белков в межклеточное пространство. А это плохо… В пульпозном ядре диска нет своих (резидентных) специализированных макрофагов. Клетки сами по мере своей возможности занимаются фагоцитозом, что критически влияет на скорость обнаружения и захват мертвых клеток. Апоптоз быстро превращается в некроз, что может спровоцировать воспаление.
Смерть клетки пироптозом – воспалительная смерть – это разбухание с последующим ее разрывом вследствие патологических внутриклеточных процессов из-за вхождения большого количества воды через образовавшиеся патологические поры в мембране. И с дальнейшим вовлечением соседних клеток, отвечающих воспалительной реакцией на это событие. Большое количество одновременно погибших клеток в результате пироптоза может спровоцировать масштабное воспаление в диске. Как правило, пироптоз вызван или критическим скисанием диска (водители – дальнобойщики) или оксидативным стрессом в результате механической перегрузки диска. Просидел 8 часов в офисе, диски поскисали, затем поехал в тренажерный зал и потаскал штангу. Убойная комбинация: «скисший и просевший диск плюс повышенная механическая нагрузка» – комбо! Критически низкое pH, повышенное количество перекисей в цитозоле, компрессия, превышающая нагрузку в 1,3 MPa и активация интегринов-бета 1 через оксидоредуктазу (LOX) приводят к активации сигнального пути p38 MAPK, вследствие чего резко увеличивается активация синтеза металлопротеиназ (MMP-3/13), аггреканаз (ADAMTS-4/5), белков старения (р21, р53 и р16) и смерти (каспаза 3,9). Можно сразу в пироптоз! Клетки хлопают как попкорн.